Volume of distribution
The volume of distribution (VD) , also known as apparent volume of distribution, is a pharmacological term used to quantify the distribution of a medication between plasma and the rest of the body after oral or parenteral dosing. It is defined as the theoretical volume in which the total amount of drug would need to be uniformly distributed to produce the desired blood concentration of a drug.[1] [2]
Volume of distribution may be increased by renal failure (due to fluid retention) and liver failure (due to altered body fluid and plasma protein binding). Conversely it may be decreased in dehydration.
The initial volume of distribution describes blood concentrations prior to attaining the apparent volume of distribution and uses the same formula.
Contents |
Equations
The volume of distribution is given by the following equation:
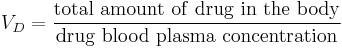
Therefore the dose required to give a certain plasma concentration can be determined if the VD for that drug is known. The VD is not a physiologic value; it is more a reflection of how a drug will distribute throughout the body depending on several physicochemical properties, e.g. solubility, charge, size, etc.
The units for Volume of Distribution are typically reported in (ml or liter)/kg body weight. The fact that VD is a ratio of a theoretical volume to a fixed unit of body weight explains why the VD for children is typically higher than that for adults, even though children are smaller and weigh less. As body composition changes with age, VD decreases.
The VD may also be used to determine how readily a drug will displace into the body tissue compartments relative to the blood:
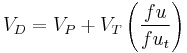
Where:
- VP = plasma volume
- VT = apparent tissue volume
- fu = fraction unbound in plasma
- fuT = fraction unbound in tissue
Examples
Understanding volume of distribution
If you administer a dose D of a drug intravenously in one go (IV-bolus), you would naively expect it to have an immediate blood concentration 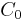 which directly corresponds to the amount of blood contained in the body
which directly corresponds to the amount of blood contained in the body 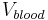 . Mathematically this would be:
. Mathematically this would be:
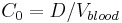
But this is generally not what happens. Instead you observe that the drug has distributed out into some other volume (read organs/tissue). So probably the first question you want to ask is how much of it is no longer in the blood stream? The volume of distribution 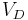 quantifies just that by specifying how big a volume you would need in order to observe the blood concentration actually measured.
quantifies just that by specifying how big a volume you would need in order to observe the blood concentration actually measured.
A practical example for a simple case (mono-compartmental) would be to administer D=8 mg/kg to a human. A human has a blood volume of around 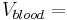 0.08 l/kg [3]. This gives a
0.08 l/kg [3]. This gives a 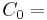 100 µg/ml if the drug stays in the blood stream only, and thus its volume of distribution is the same as that is
100 µg/ml if the drug stays in the blood stream only, and thus its volume of distribution is the same as that is 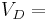 0.08 l/kg. If the drug distributes into all body water the volume of distribution would increase to approximately 0.57 l/kg [4]
0.08 l/kg. If the drug distributes into all body water the volume of distribution would increase to approximately 0.57 l/kg [4]
If the drug readily diffuses into the body fat the volume of distribution may increase dramatically, an example is chloroquine which has a 250-302 l/kg [5]
In the simple mono-compartmental case the volume of distribution is defined as: 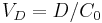 , where the in practice is an extrapolated concentration at time=0 from the first early plasma concentrations after an IV-bolus administration (generally taken around 5min - 30min after giving the drug).
, where the in practice is an extrapolated concentration at time=0 from the first early plasma concentrations after an IV-bolus administration (generally taken around 5min - 30min after giving the drug).
Further reading:
Table of volume of distribution for drugs
| Drug | VD | Comments |
| Warfarin | 8L | Reflects a high degree of plasma protein binding. |
| Theophylline, Ethanol | 30L | Represents distribution in total body water. |
| Chloroquine | 15000L | Shows highly lipophilic molecules which sequester into total body fat. |
| NXY-059 | 8L | Highly-charged hydrophilic molecule. |
Sample values and equations
| Characteristic | Description | Example value | Abbreviation(s) | Formula |
|---|---|---|---|---|
| Dose | Loading dose (LD), or steady state / maintenance dose (MD). | 500 mg | 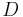 |
design parameter |
| τ | Dosing interval. | 24 h | 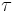 |
design parameter |
| Volume of distribution | The apparent volume in which a drug is distributed immediately after it has been injected intravenously and equilibrated between plasma and the surrounding tissues. | 6.0 L | 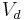 |
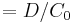 |
| Concentration | Initial or steady-state concentration of drug in plasma. | 83.3 µg/mL | 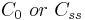 |
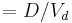 |
| Biological half-life | The time required for the concentration of the drug to reach half of its original value. | 12 h | 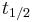 |
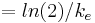 |
| Elimination rate constant | The rate at which drugs are removed from the body. | 0.0578 h-1 | 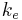 |
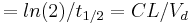 |
| Elimination rate | Rate of infusion required to balance elimination. | 50 mg/h | 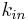 |
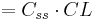 |
| Area under the curve | The integral of the concentration-time curve (after a single dose or in steady state). | 1320 µg/mL×h | 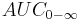 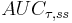 |
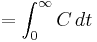 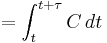 |
| Clearance | The volume of plasma cleared of the drug per unit time. | 0.38 L/h | 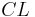 |
 |
| Bioavailability | The fraction of drug that is absorbed. | 0.8 | 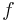 |
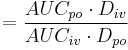 |
| Cmax | The peak plasma concentration of a drug after oral administration. | 60.9 µg/mL | 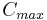 |
direct measurement |
| tmax | Time to reach Cmax. | 3.9 h | 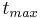 |
direct measurement |
| Cmin | The lowest (trough) concentration that a drug reaches before the next dose is administered. | 27.7 µg/mL | 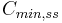 |
direct measurement |
| Fluctuation | Peak trough fluctuation within one dosing interval at steady state | 41.8 % | 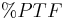 |
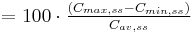 where 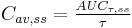 |
| edit | ||||
References
- ^ http://www.merckmanuals.com/professional/sec21/ch324/ch324d.html
- ^ "vetmed.vt.edu". http://cpharm.vetmed.vt.edu/VM8314/VM8314PharmaModeling.htm.
- ^ Alberts, Bruce (2005). "Leukocyte functions and percentage breakdown". Molecular Biology of the Cell. NCBI Bookshelf. http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Search&db=books&doptcmdl=GenBookHL&rid=mboc4.table.4143. Retrieved 2007-04-14.
- ^ Guyton, Arthur C. (1976). Textbook of Medical Physiology (5th ed.). Philadelphia: W.B. Saunders. p. 424. ISBN 0-7216-4393-0.
- ^ Wetsteyn JC (1995). "The pharmacokinetics of three multiple dose regimens of chloroquine: implications for malaria chemoprophylaxis". Br J Clinical Pharmacology 39 (6): 696–9. PMID 7654492.
External links
- Tutorial on volume of distribution
- Overview at icp.org.nz
- Overview at cornell.edu
- Overview at stanford.edu
- Overview at boomer.org
|
|||||||||||||||||